Болезнь Гоше: причины возникновения и основные симптомы, способы лечения заболевания
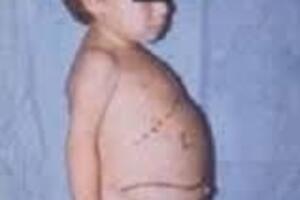
Представляет собой генетическое заболевание, проявляющееся нарушением липидного обмена, недостаточностью лизосомальных ферментов и накоплением гликолипидов в клеточных структурах.
Причины
Глюкозилцерамидный сфинголипидоз – это самая частая форма из всех наследственных ферментопатий. Причиной его развития считается дефект гена GBA, который кодирует фермент лизосом бета-глюкозидазу, который отвечает за расщепление липидов. Наследование болезни происходит аутосомно-рецессивному типу, для формирования ферментопатии необходимо присутствие пары измененных генов: один – от матери, а другой – от отца. В супружеской паре, где оба родителя являются носителями мутации, вероятность рождения больного ребенка достигает 25%. Риск передачи одного дефектного гена, то есть риск носительства без развития болезни в таких семьях составляет 50%.
Симптомы
По возрасту манифестации и особенностям клинической картины различают три типа болезни. Первый тип является самым распространенным и отличается хроническим характером течения. Симптомы недуга появляются примерно к 30 или 40 годам, реже дебютируют в детском возрасте. Увеличение в размерах печени и селезенки отмечается у ребенка сразу после рождения, но клинические симптомы заболевания проявляется позже. Первыми признаками недуга являются анемия и повышенная кровоточивость. Ингибирование работы системы кроветворения сопровождается снижением уровня гемоглобина и тромбоцитов. Изменения со стороны опорно-двигательного аппарата проявляются болями в костях и суставах, частыми переломами и деформациями. У взрослых появляется гиперпигментация на лице и ногах, в области поражения кожа темнеет, приобретает оттенок от желтоватого до желто-коричневого. Возможно появление плоских красных пятен с типичной локализацией в области вокруг глаз.
Второй тип болезни встречается достаточно редко, развивается в промежутке от рождения до полутора лет. Дебют заболевания происходит в первые три месяца жизни. Заболевание характеризуется стремительным течением, недостаточным откликом на лечение. Сначала у больного возникают неврологические расстройства, спровоцированные скоплением клеток Гоше в центральной нервной системе. Такие малыши слабо кричат и вяло сосут. У них нарушен глотательный рефлекс, нередко выявляется нарушение циклов дыхания. Наблюдается заметная задержка психического и физического развития. На начальной стадии заболевания мышечный тонус снижен, через 9-12 месяцев после дебюта возникает гипертонус, особенно в мышцах шеи и конечностях. У ребенка появляются судороги, косоглазие, спастический паралич. Печень и селезенка увеличены.
Третий тип – ювенильный или подострый нейропатический. Первые признаки недуга – это увеличение в размерах селезенки и печени, выявляются у малыша с 2 до 3 лет. Клиническая картина полностью разворачивается в период с 6 до 15 лет. Клинически заболевание проявляется поражением центральной нервной системы развитием гипертонуса мышц, параличей спастического типа, косоглазия, непроизвольных спазмов, судорог, затрудненного дыхания с трудностью вдоха, проблемами при глотании. У больного может отмечаться снижение интеллектуальных функций, несформированность речи и письма, эмоциональная неустойчивость и психоз. Дети отстают в половом развитии. Даная форма заболевания отличается прогрессирующим течением.
Диагностика
Больному проводится физикальный осмотр и сбор анамнеза в процессе которого выясняется наличие болезни Гоше у родственников. При осмотре выявляются типичные признаки: низкий рост, патологии костей, неврологические симптомы, геморрагический синдром, гиперпигментация кожи. Для подтверждения диагноза больному назначается клиническое, биохимическое исследование крови, ферментный анализ клеток, морфологическое изучение костного мозга, исследование структуры костной ткани, а также ультразвуковое исследование печени и селезенки.
ДНК-диагностика – это необязательная процедура. Его проводят в случае необходимости подтверждения мутации в гене GBA.
Лечение
Специализированная помощь больным с первым и третьим типом болезни направлена на устранение симптомов и компенсацию первичного генетического дефекта, и повышение количества недостающего фермента, усиление катаболизма гликосфинголипидов. Основным методом лечения является пожизненная ферментная заместительная терапия с применением рекомбинантной глюкоцереброзидазы. Эффективность лечения достаточно высока – симптомы полностью купируются, а качество жизни больных повышается.
Профилактика
Профилактика проводится во время планирования беременности и на ее начальных сроках. Медико-генетическое консультирование рекомендуется семьям, имеющим близких родственников с данной патологией.
Связанные лекарства:
Информация является обобщающей и не может быть использована для лечения, без рекомендации врача.

