Болезнь Штаргардта: причины возникновения и основные симптомы, способы лечения заболевания
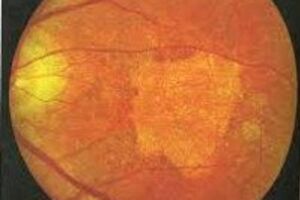
Наследственное заболевание сетчатки глаза, проявляющееся развитием дистрофических изменений в ее макулярной зоне, что сопровождается потерей центрального зрения.
Причины
Передача заболевания происходит преимущественно по аутосомно-рецессивному типу, что указывает на то, что наследование недуга несцеплено с полом и далеко не всегда передается ребенку. Сегодня выявлены случаи наследования мутированного гена и по доминантному типу. При доминантном типе наследования поврежденного гена, отвечающего за синтез белка-транспортера АТФ, заболевание отличается более легким течением и только в редких случаях приводит к инвалидизации больного.
При таком типе наследования большинство рецепторных клеток макулы желтого пятна глазного дна сохраняют свою функциональность. У лиц с доминантным типом наследования заболевание обусловлено возникновением минимальных нарушений, благодаря чему некоторые пациенты способы даже водить автотранспорт.
Основной причиной дегенерации клеток макулы является дефицит энергии, возникающий на фоне синтеза неполноценного белка, который отвечает за транспортировку молекул АТФ сквозь мембрану клеток желтого пятна, представляющего собой центр сетчатки глаза, в котором фокусируется графическое и цветное изображение. Желтое пятно лишено кровеносных сосудов, а питание клеток-колбочек происходит посредством белков-переносчиков АТФ из близлежащей сосудистой оболочки. Белки переносят сквозь мембрану в средину клеток-колбочек молекулы АТФ.
В норме родопсин фоторецепторов поглощает фотон света, превращаясь в транс-ретиналь и опсин. На следующем этапе транс-ретиналь под воздействием энергии АТФ, которую дают белки-переносчики, превращается в ретиналь, соединяющийся с опсином. Так восстанавливается родопсин. При аберрации гена формируется неполноценный белок-переносчик. Вследствие этого происходит нарушение восстановления родопсина и накопление транс-ретиналя, который трансформируется в липофусцин, оказывающий прямое токсическое воздействие на клетки-колбочки.
Симптомы
Дебют заболевания происходит в возрасте от 6 до 7 лет. Вне зависимости от типа наследования у больного наблюдается формирование центральной скотомы. В случае легкого течения скотомы относительны: такие больные видят яркие цвета и способны различать контуры предметов, но при этом они плохо различают объекты, обладающие недостаточно выраженным окрасом.
У большинства больных выявляется нарушение цветового зрения по типу красно-зеленой дисхромазии, обусловленной тем, что человек различает светло-зеленый цвет как темно-красный. У некоторых больных, сохраняется нормальное восприятие цветовой гаммы.
На начальном этапе заболевания у пациента не отмечается изменения границ периферического зрения, при прогрессировании центральной скотомы границы зрения расширяются, что с течением времени приводит к слепоте. Параллельно с возникновением выпадения центрального зрения отмечается снижение его остроты. На заключительной стадии болезни Штаргардта происходит атрофия зрительного нерва, в результате чего пациент полностью теряет зрение.
Диагностика
Возникновение симптомов заболевания в детском возрасте – основной из критериев, который обязательно учитывается при постановке диагноза. Для подтверждения диагноза больному назначается проведение офтальмоскопии, периметрии, флуоресцентной ангиографии и молекулярно-генетического анализа.
Лечение
На данный момент не разработано этиологического лечения болезни Штаргардта. В качестве общего вспомогательного лечения применяются парабульбарные инъекций таурина и антиоксидантов, введение сосудорасширяющих средств, стероидных препаратов. Также больному может быть назначена витаминотерапия, направленная на укрепление сосудов и улучшение кровоснабжения. В тяжелых случаях может потребоваться проведение реваскуляризации сетчатки путем трансплантации в зону желтого пятна пучка мышечных волокон.
Профилактика
На данный момент не разработано эффективных методов профилактики развития болезни Штаргардта. Пары с неблагоприятным семенным анамнезом на стадии планировании беременности должны посетить генетика.
Связанные лекарства:
Информация является обобщающей и не может быть использована для лечения, без рекомендации врача.

